Dear All,
first congrats for the great tool you’ve developed.
I’ve undertaken an n-integration analysis of three omics (metabolomics, mrna and mirna) from a series of rat samples with block.spslsda() function in mix omics [also undertaking the tuning with tune.block.spslsa()].
Now I have some questions if possible with regards of the outputs obtained:
- As the omics data was already log-transformed, normalised (array wise) and filtered, was I right assigning ‘scale=F’ as parameter for both block.splsa(), perf() and tune.block.splsda() throughout the analysis process?
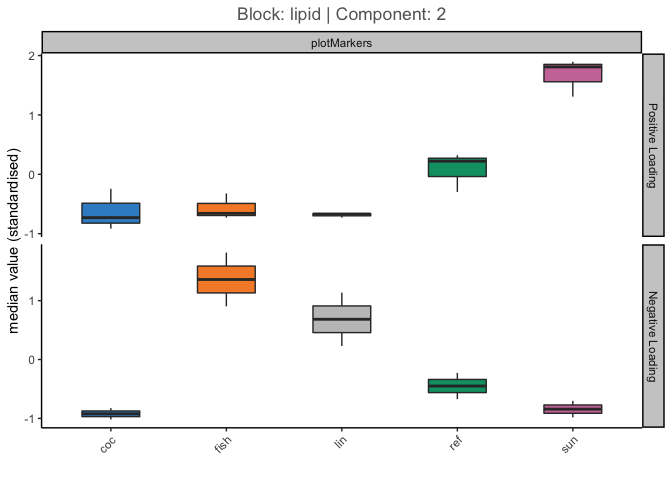
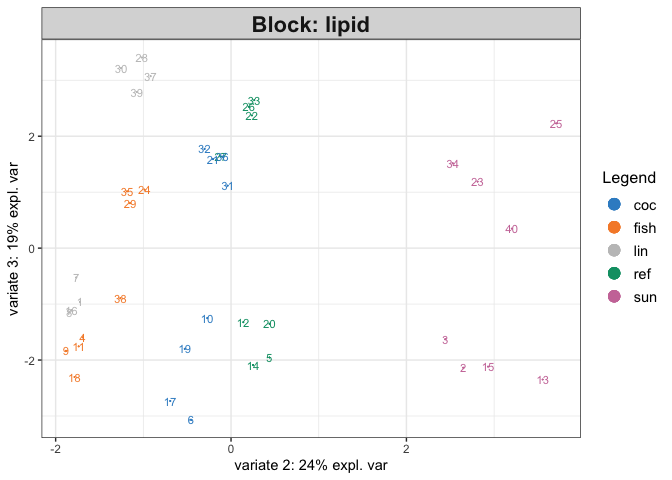
- Circos Plot obtained and hereby shown:
From Gonzalez et al 2012 I could not find a complete answer to how to interpret the 4 group lines (I’ve highlighted them in Red in the screen).
Are they describing the raw data in terms of different expression between the 4 groups of my Y (4 different brain areas) e.g. meaning that in the red highlighted area the mRNA in the blue group (Cervelletto) are underexpressed with respect of the remnant 3 groups? Plus, are the selected features shown in the circos plot from just the 1st component or from all the component selected as best by block.splsda() (4 components in my case)?
3)CIN heatmap:
Are the selected features by tune.block.splsda() shown in the heatmap from the 1st component only or from all the selected components (4 in my case). Plus, in terms of rCC analysis (as it’s not Pearson being the values in the top left scale not restricted between 0-1 right?) and in order to provide a biological interpretation of such plot, how shall I interpret the concept of correlation with respect of a specific feature selected by the algorithm? (e.g. the metabolite/genes selected are up-down correlated with respect to what? The 4 groups (Y) in my case? Or with respect of the samples?).
Or I should try to recreate the heatmap in Figure 10 of Gonzalez et al 2012 for a better biological interpreation?.


Apologies for the long question and many thanks in advance for the great support.
Best regards
Joosè